

欢迎光临吉康旅!
目前,国家加快了海外创新新药的上市步伐,但等待国外上市的新药在国内上市还有一段时间。海外上市药物一直是广大癌症患者及其家属关注的焦点。参考FDA和CFDA的官方资料,我们整理了中国最全面的未上市肿瘤药物系列。根据不同的癌症类型,我们可以为肿瘤科医生、患者及其家属提供参考!此条目介绍的是肺癌。
1、(索托拉西布)
产品名称:
制造商:安进
上市时间:2020年2月(香港)
包装规格:×240个
适应症:用于治疗接受过至少一种全身治疗的 KRAS G12c 突变非小细胞肺癌()患者。
使用方法:每日1次。
KRAS 是肿瘤的常见驱动因素,存在于近 90% 的胰腺癌、30-40% 的结肠癌和超过 30% 的肺腺癌中。然而,近 40 年来,靶向 KRAS 的小分子药物的开发并没有取得重大突破,因为 KRAS 蛋白表面没有适合小分子抑制剂结合的口袋。2019年ASCO大会上,成功破冰,一期研究成果惊艳全场,为KRAS靶向治疗带来历史性时刻。
2018年8月,该药在I期临床试验注册,不到一年就取得了良好的效果。已披露正面数据;
2019年5月1日和2019年5月23日,FDA分别批准了治疗非小细胞肺癌()和结直肠癌(CRC)的孤儿药资格;
2020年3月9日,CDE(国家食品药品监督管理局)官网更新,安进公司研发的KRAS G12C抑制剂AMG 510的临床应用由药品审评中心承担;
2020年12月9日,KRAS G12C突变二线治疗药物获得FDA突破性疗法认定,并于当月16日提交上市申请。
在2020年ASCO、WCLC和ESMO三大肿瘤学会议上发表。I期研究结果显示,KRAS突变(非小细胞肺癌)患者的总体ORR(客观缓解率)为48%,DCR(疾病控制率)为96%。在接受 960 mg AMG 510 的 13 名患者中,ORR 为 54%,DCR 为 100%。在 KRAS 突变的结直肠癌患者中,DCR 达到 79%。
2021 年 5 月 29 日,(AMG-510,商品名:)获得 FDA 的加速批准,用于治疗接受过至少一种全身治疗的 KRAS G12c 突变的非小细胞肺癌( )患者。这是全球首个针对KRAS的靶向药物,具有里程碑意义!
批准基于 100 项研究的数据。根据 100 项研究的最新数据,在中位随访 12.2 个月后,124 名既往接受过化疗和/或免疫治疗的疾病进展突变患者纳入疗效分析,共有 46 名受试者获得确认反应,其中 3 名完全反应和 43 名部分反应,达到 37.1% 的客观反应率 (ORR)。达到客观反应的中位时间为 1.4 个月,中位反应持续时间为 10 个月,43% 的患者在没有疾病进展的情况下继续治疗。疾病控制率为80.6%,中位无进展生存期(PFS)为6.8个月。
大约 3% 的结直肠癌患者有 KRAS 突变。2021年ESMO GI会议上公布的数据显示,()治疗KRAS G12C突变的结直肠癌患者,无论是继发性还是原发性耐药,ORR为7.1%,DCR为73. 8%,中位 PFS 为 4 个月。
2、(卡马替尼)
产品名称:
制造商:(诺华)
上市时间:2020 年 5 月(美国)
包装规格:56个/瓶
适应症:突变型肺癌,是突变疗效最高的药物之一。
使用方法:每日2次。
41% 的患者之前经历过两种以上的治疗方案。中位反应持续持续时间分别为 12.6 个月和 9.7 个月。结果表明,无论患者是否接受过前期治疗(一线和后期),均取得了显着的治疗效果。但是无进展生存期(PFS)并不是特别长,只有4个月左右,所以我们也期待更多的药物数据。
ASCO 报告了 160 名患者,包括未治疗的患者(队列 5b 和 7)和先前接受过 1L 或 2L 治疗的患者(扩展队列 6 和队列 4)。扩展队列 7 的 ORR 为 65.6%,中位数PFS 为 10.8 个月,中位 OS 未成熟。队列 5b,ORR 为 67.9%,OS 为 20.8 个月,中位 PFS 为 12. 4个月。卡马替尼治疗跳过突变@>6个月的患者ORR为40.6%,OS为13.。因此卡马替尼一线治疗可以获益更多。
此外,卡马替尼还表现出良好的颅内反应。在 mono-1 试验中,13 名有神经放射学数据的患者中,92% 有颅内转移,54% 表现出良好的颅内反应(31% 表现出完全缓解)。在所有接受卡马替尼治疗的患者中,最常见的 AE(任何原因;≥20%)是外周水肿、恶心、呕吐、血清肌酐升高、呼吸困难、疲劳和食欲不振。
3、特泊替尼(®)
产品名称:
制造商:默克
上市时间:2020 年 2 月 3 日(FDA)
包装尺寸:×60
适应症:用于治疗不可切除、MET外显子14跳跃突变晚期或复发性非小细胞肺癌患者。
使用方法:每日一次,饭后口服
()是一种口服、高选择性IB型MET抑制剂,已在美国和日本获批。2021 年 2 月 3 日,FDA 加速批准上市,用于治疗携带 MET 外显子 14 () 跳跃突变的转移性非小细胞肺癌 () 患者。NCCN V4指南也将升级为晚期突变的一线优先推荐。
该药物的批准是基于 II 期试验的结果,这是一项前瞻性、非随机、开放标签的 II 期研究,对 152 名晚期或转移性 MET 外显子 14 外显子跳跃患者进行了每日给药 (/d)。独立审查委员会 (BIRC) 确定 ORR 为 43% (95%CI, 32-56), mDOR 10.8 个月 (95%CI, 6.9 个月-NE );治疗患者 8.5 个月的 ORR 为 43% (95%CI, 33-55), .1 个月 (95%CI, 9.5-1))。最常见的不良反应(任何原因;≥20%)是水肿、疲劳、恶心、腹泻、血清肌酐升高、肌肉骨骼疼痛和呼吸困难。
在 2021 年 ELCC 大会上更新了 II 期研究中 A 组(外显子跳跃突变人群)的疗效数据。在 152 名患者中,治疗的 ORR 达到 44.7%,初治患者和接受过治疗的患者的 ORR 相似(均为 44.9%)。总人群的中位 DOR 为 11.1 个月,初治人群和接受过治疗人群的数据相似(10.8 个月和 11.1 个月);中位 PFS 为 8.9 个月(初始治疗 8.5 个月,治疗后 10.9 个月)。颅内转移患者的ORR为47.8%,中位DOR为9.5个月,中位PFS为9.5个月。在安全方面,
4、 (-vmjw)
通用名称:-vmjw
产品名称:
全名:,-vmjw
制造商: , Inc.
包装规格:350 mg/7 mL(50 mg/mL)/瓶/盒,无菌、无防腐剂、无色至淡黄色静脉输液溶液。
适应症:
一种针对 EGF 受体和 MET 受体的双特异性抗体,适用于治疗患有表皮生长因子受体 (EGFR) 外显子 20 插入突变的局部晚期或转移性非小细胞肺癌 () 成年患者,FDA 批准的测试,其疾病在基于铂的药物上或之后发展了化学疗法。
剂量:
推荐剂量基于基线体重,并在稀释后作为静脉输注给药。
按照建议服用处方药。在第 1 周和第 2 周通过外周线给药。每周给药 4 周,从第 1 周的第 1 天和第 2 天推注初始剂量开始,然后每 2 天一次,持续数周。通过静脉输注稀释。
不良反应:
最常见的不良反应(≥20%)是皮疹、IRR、甲沟炎、肌肉骨骼疼痛、呼吸困难、恶心、疲劳、水肿、口腔炎、咳嗽、便秘和呕吐。最常见的 3 级或 4 级实验室异常(≥2%)是淋巴细胞减少、白蛋白减少、磷酸盐减少、钾减少、碱性磷酸酶增加、葡萄糖增加、γ-谷氨酰转移酶增加和钠减少。
禁忌症:无。
防范措施:
输液相关反应 (IRR):在出现 IRR 的第一个迹象时中断输液。根据严重程度降低输注速度或永久停药。
间质性肺病 (ILD)/肺炎:监测表明 ILD 的新症状或恶化症状。立即停止疑似 ILD/肺炎的住院患者,如果确诊 ILD/肺炎,则永久停止治疗。
皮肤不良反应:可能引起皮疹,包括痤疮样皮炎和中毒性表皮坏死松解症。根据严重程度扣留、减少剂量或永久停药。
眼部毒性: 立即将眼部症状恶化的患者转诊给眼科医生。根据严重程度扣留,减少剂量或永久停药。
胚胎-胎儿毒性:可能对胎儿造成伤害。建议对胎儿有潜在生殖风险的妇女使用有效的避孕措施。
在特定人群中使用
哺乳期:不建议母乳喂养。
贮存:
在 2°C 至 8°C(36°F 至 46°F)的冰箱中储存在原始纸箱中以避光。
机制:
-vmjw 是一种双特异性抗体,可与 EGFR 和 MET 的细胞外结构域结合。
在体外和体内研究中,-vmjw 能够通过阻断配体结合和破坏外显子 20 插入突变模型中的 EGFR 和 MET 来破坏 EGFR 和 MET 信号传导功能。肿瘤细胞表面存在 EGFR 和 MET 也使这些细胞能够分别通过抗体依赖性细胞毒性 (ADCC) 和光细胞作用机制被免疫效应细胞(如自然杀伤细胞和巨噬细胞)破坏。
安全性和有效性:
在一项对 81 名患有非小细胞肺癌和 EGFR 外显子 20 插入突变的患者进行的研究中,研究人员评估了这些患者的疗效,这些患者在铂类化疗后都出现了晚期疾病进展。研究结果显示,整体反应率为40%;中位响应时间为 11.1 个月;63% 的患者有 6 个月或更长时间的反应时间。
临床数据:
一、2021年5月21日,美国FDA加快批准强生公司研发生产的EGFR/c-Met双抗体(-vmjw, JNJ-6732),用于治疗患有以下疾病的患者)铂类化疗后的晚期化疗 EGFR 外显子 20 插入突变的转移性非小细胞肺癌 () 患者。这是 FDA 批准的首个针对此类突变的药物。除了治疗,在解决奥希替尼方面耐药性和全面覆盖EGFR靶点也很能干!
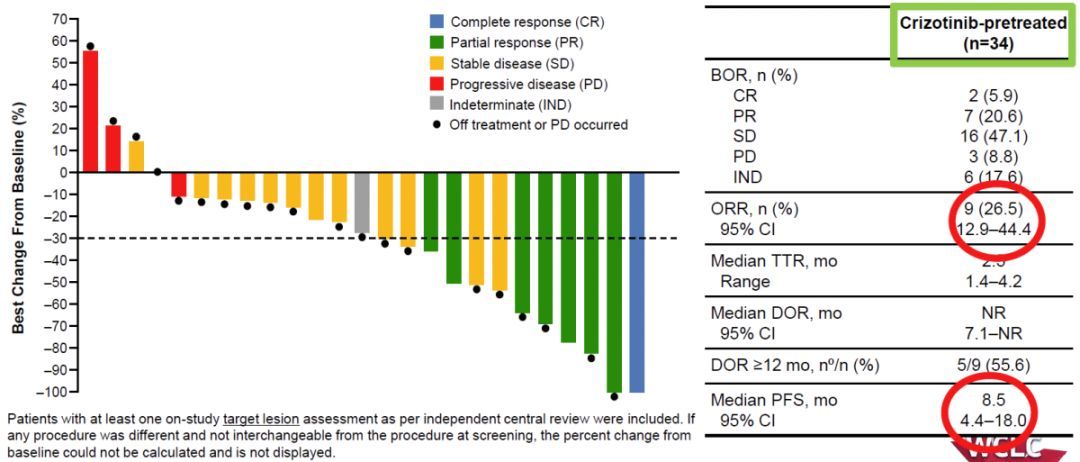
该批准基于 I 期研究单药治疗队列的阳性结果,该研究在 2020 年 WCLC 会议上更新,81 名 EGFR 阳性肺癌患者曾接受过一线铂类化疗,所有这些患者疾病进展后接受治疗。体重客观缓解率(ORR)为40%,其中完全缓解率为4%,部分缓解率为36%,缓解DoR的中位时间为11.1个月。临床获益率(定义为至少两次疾病评估的完全或部分反应或疾病稳定)为 74%。
二、在中国启动EGFR/MET双抗体(研发代码:JNJ-)的3期临床试验,评估与第三代EGFR-TKI药物联合一线治疗EGFR突变的局部晚期或转移性非小细胞肺癌 细胞肺癌的疗效( )。
三、 2020年3月,杨森药业宣布美国食品药品监督管理局(FDA)授予JNJ-(JNJ-6372) )用于治疗表皮生长因子受体转移性非- 小细胞肺癌 (EGFR) 外显子 20 插入突变在基于铂的化疗期间或之后疾病进展的患者中。肺癌治疗领域的里程碑。
突破性疗法的认定是基于 1 期、首次人体、开放标签、多中心试验的数据 ( )。该试验评估了 JNJ-6372 单药治疗和联合使用新型第三代 EGFR TKI 在成人晚期疾病中的安全性、药代动力学和初步疗效。
5、(奈昔单抗)
产品名称:
制造商:礼来公司
上市时间:2015 年 11 月 24 日(FDA)
包装规格:/50ml
适应症:与吉西他滨、顺铂联用,用于转移性鳞状非小细胞肺癌患者的一线治疗。不适合治疗非鳞状非小细胞肺癌。
使用方法:推荐剂量为(绝对剂量),每 3 周 1 个疗程,第 1 天和第 8 天静脉滴注超过 60 分钟,在吉西他滨和顺铂之前,持续至疾病进展或出现不可接受的毒性。.
最常见的肺癌类型是非小细胞肺癌(),其中鳞状细胞癌约占 30%。肺鳞癌目前以放化疗为主,靶向药物较少。( ) 是一种重组人源 IgG1 单克隆抗体,可与人表皮生长因子受体 (EGFR) 结合,从而阻断 EGFR 与其配体的结合。
在体外,EGFR 内化和降解在 EGFR 表达细胞中被诱导并引发抗体依赖性细胞毒性 (ADCC)。使用人类肿瘤异种移植模型(包括非小细胞肺癌模型)的体内研究发现,与单独使用吉西他滨和顺铂治疗相比,给予小鼠可提高吉西他滨和顺铂组合的抗肿瘤活性。
礼来最初在非小细胞肺癌 () 中测试了两个 III 期临床对,一个在鳞状细胞群中进行 III 期试验,另一个在非鳞状细胞群中进行 III 期试验。2011 年初,BMS 中止了试验,因为治疗组中的一些患者出现了血栓栓塞。鳞状上皮患者的试验被允许继续进行,BMS 后来终止了合作并归还了全部权利。
2015年11月24日,美国食品药品监督管理局(FDA)批准()与吉西他滨和顺铂联合用于治疗晚期鳞状非小细胞肺癌()。
该批准基于转移性鳞状肺癌人群的一项随机 III 期研究,其中患者随机接受吉西他滨 + 顺铂 + mAb(研究组,n=545)/安慰剂(对照组)组,n =548)。给药方案为1、第8天,每3周一次。两组吉西他滨给药方案为1、第8天 /m2,第 1 天以 75 mg/m2 的剂量给予顺铂。研究组中有反应的患者继续接受单药 mAb。
两组患者的基本特征相似。中位年龄为 62 岁,大多数患者是白人 (84%),绝大多数患者有吸烟史 (91%),最常见的转移是肺部 (83%)。研究的主要终点是 OS,次要终点是 PFS 和客观缓解率 (ORR)。
结果显示,经过近25个月的随访,研究组与对照组(下同)的中位OS分别为11.5个月vs.9.9个月(HR =0.84; 95%CI 0.74-0.96;P=0.012). 1 年 OS 率:48% vs. 43 %;2 年 OS 率:20% 对 17%。
中位 PFS:5.7 个月与 5.5 个月(HR=0.85;95%CI 0.74-0.98;P =0.02);6 个月 PFS 率:45% 对 37%。
ORR:31% vs. 29% (P=0.40);疾病控制率(ORR+疾病稳定):82% vs. 77% (P=0.043);治疗失败的中位时间:4.3 个月 vs. 3.6 个月。
3 级或更高级别不良事件 (AE) 的发生率分别为 72% 和 62%。研究组中 3 级及以上的 AE 显着增加为:低镁血症(9% 对 1%)、皮疹(4% 对 1%)。
严重 AE 率:48% 对 38%。导致治疗中止的 AE 发生率为 31% 和 25%。AE 后的死亡率:12% 对 11%。
分子标志物探查结果显示,Ⅳ期肺鳞癌患者有EGFR表达(免疫组化方法检测至少1个阳性细胞,EGFR>0),但无EGFR表达(EGFR=0)患者亚组没有显着的临床益处。
目前,晚期鳞状细胞癌的治疗选择非常有限。联合治疗可提高鳞癌患者的生存率,对未来的肿瘤治疗具有重要意义。
免责声明:本资料所含信息仅供参考,请听从您的医生或其他医疗保健专业人士的建议或指导。
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
