

欢迎光临吉康旅!
肺癌是一种高发癌症,其类型通常有腺癌、鳞癌、大细胞癌和小细胞癌。其中,肺腺癌占比最高,对应的靶向药物最多,主要是EGFR-TKI类、ALK-TKI类、ROS-TKI类药物。随着众多临床研究的开展,肺癌靶向药物的耐药机制已较为明确。
EGFR-TKI 耐药
在肺腺癌的治疗中,使用最多的药物是EGFR抑制剂-TKI(酪氨酸激酶抑制剂),如第一代药物:吉非替尼、厄洛替尼,第二代药物:阿法替尼、达克替尼、第三代药物:奥希替尼、阿美替尼、福美替尼。治疗过程中,EGFR自身产生继发性耐药突变(如突变),以及RAS、BRAF等基因突变、HER2、MET等基因扩增、ALK、RET基因重排或融合, 可导致患者对 TKI 产生耐药性。
奥希替尼是目前最有效的 TKI 药物。在EGFR-lung其他学者的文章中,对奥希替尼的耐药机制进行了梳理,分类为:
EGFR 依赖:
EGFR突变:例如该突变是奥希替尼的典型耐药突变位点。与一线治疗相比,二线治疗可能会产生更多的耐药突变位点。此外,20外显子存在溶剂前沿突变,涉及铰链区的突变可能会干扰奥希替尼与靶蛋白的结合。
EGFR-TKI 敏感(绿色)/耐药(橙色)突变
EGFR扩增:也有文献报道,经过3代EGFR-TKI药物(如奥希替尼)耐药后发现EGFR扩增存在野生型。
EGFR 独立:
MET扩增:MET扩增的耐药机制是MET结合,绕过EGFR激活下游PIK3/AKT介导的信号通路,促进肿瘤细胞增殖,抑制细胞凋亡。 MET扩增可引起EGFR-TKI耐药,但此时EGFR通路仍处于活跃状态。为克服耐药性,可在原有EGFR-TKI基础上联合MET通路抑制剂。
其他获得性扩增:如HER2、KRAS、BRAF、扩增,其中值得注意的是,HER2扩增往往与突变互斥,不会共存。
获得性突变:HER2、FGFR、BRAF、KRAS、MET等基因的点突变也是EGFR-TKI耐药的常见原因。
细胞周期基因的变化:细胞周期相关基因CDK4、CDK6、的扩增或突变也会引起耐药性。
组织学转化:使用第一代 EGFR-TKI 后,14% 的 EGFR 突变肺癌转化为小细胞肺癌 (SCLC)。无论一线或二线治疗如何,使用第三代 EGFR-TKI 后发生 SCLC 转化的患者比例相似(4-15%)。近 15% 的患者还转化为肺鳞状细胞癌。这些组织学转变会导致耐药性。
基因融合/重排:一些比较少见的基因融合如RET(RET-ERC1、-RET和-RET)、BRAF(AGK-BRAF、-BRAF、-BRAF和-BRAF)、NTRK(TPM3-)、 ROS1 (GOPC–ROS1), and ALK(–ALK), 是癌症驱动因子,也与奥希替尼耐药有关。
未知原因:除上述原因外,还有大量耐药原因无法确定,需要进一步研究。
总的来说,奥希替尼一线和二线治疗各部分的耐药机制比例略有不同,大致接近,但二线治疗往往会产生更多的耐药突变。
奥希替尼一线(b)和二线(a))治疗耐药因素
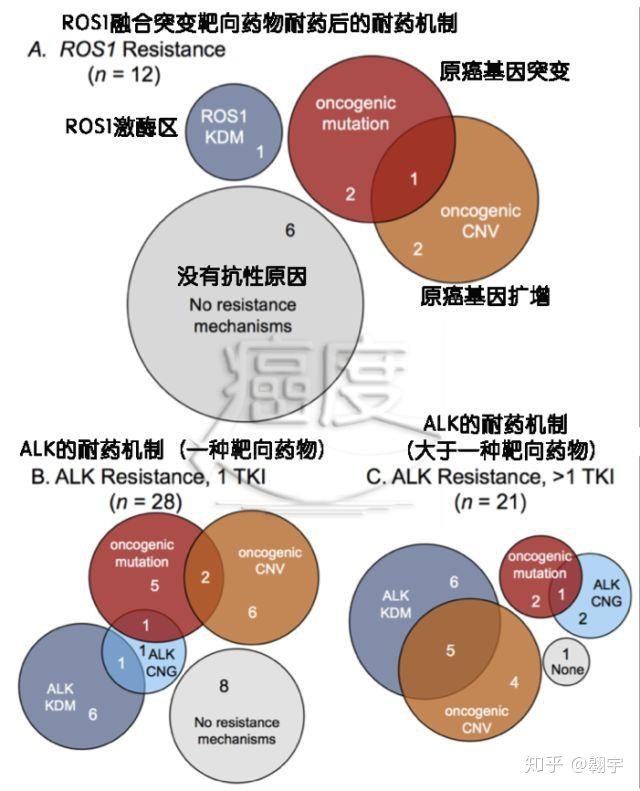
ALK-TKI 耐药
ALK是胰岛素样受体酪氨酸激酶家族(RTK)的成员,ALK基因重排是驱动基因。 ALK融合阳性已被定义为一种特殊的亚型,其中以EML4-ALK最为常见,除ALK外,还有25种融合型。相关靶向药物中,第一代为克唑替尼,第二代为色瑞替尼、艾乐替尼、布加替尼,第三代为劳拉替尼。
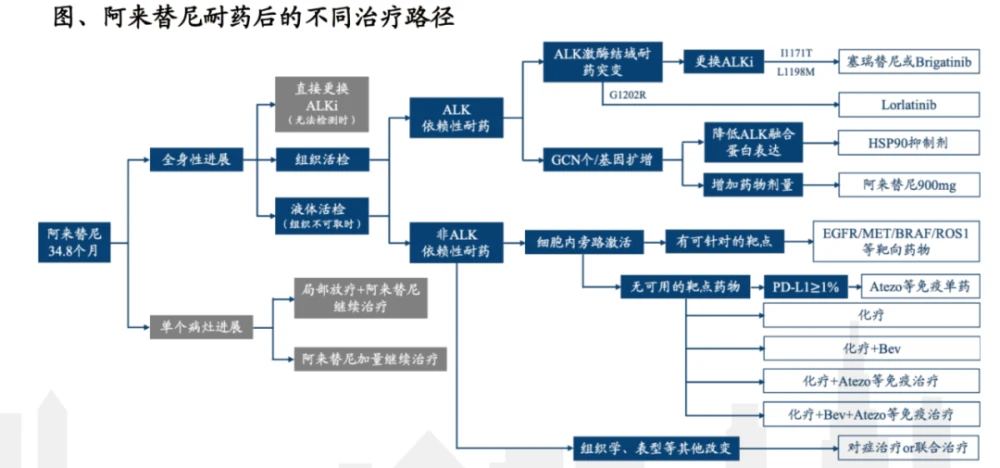
对于晚期 ALK 重排,二代 ALK-TKI 作为一线标准被广泛接受,因为二代 TKI 改善了 PFS、CNS 活性,并且比克唑替尼具有更有利的毒性特征。在第二代 ALK-TKI 疾病进展后,ALK 激酶结构域中的外显子获得性耐药突变更为常见,并在超过 50% 的病例中诱导耐药,而在使用克唑替尼时,这一比例为 20% 至 30%。此外,继发性 ALK 激酶突变的谱系因先前的 TKI 暴露而异。对于第二代 ALK-TKI 色瑞替尼、艾乐替尼和布加替尼,ALK 是疾病进展中最常见的突变;然而,携带这种突变的肿瘤仍然对第三代 TKI 劳拉替尼敏感。事实上,当出现 ALK-TKI 耐药突变时,可以采用量身定制的后续治疗方案,具体取决于先前的 TKI 暴露和特异性耐药突变。 ALK 是一项由美国国家癌症研究所赞助的研究。本试验旨在研究二代ALK-TKI治疗后疾病进展患者如何根据生物标志物选择治疗方案。
脱靶通路激活并不是二代ALK-TKI耐药的常见原因,但在某些情况下,脱靶通路激活引起的耐药可导致靶向治疗,如EGFR通路激活和MET扩增。已经报道了组织学转化(例如,上皮-间质转化、鳞状细胞癌和由于小细胞转化引起的耐药性),但仅限于个别病例。后来,最近的一项 III 期试验表明,对于既往未接受过全身治疗的 ALK 重排患者,劳拉替尼比克唑替尼更有效,导致美国食品和药物管理局 (FDA) 批准了这一适应症。一线劳拉替尼的使用仍有待观察,但已经报道了对二线和三线劳拉替尼治疗耐药的独特机制,包括新型化合物 ALK 突变和 NF2 功能丧失突变。
ROS1-TKI 抗性
ROS1基因突变,包括融合、突变和扩增,会导致ROS1酪氨酸激酶结构域和下游信号通路的持续激活,从而导致肿瘤进展。发生。融合是 ROS1 的主要变体类型,已确定至少 55 个融合伙伴。 ROS1-TKI药物包括克唑替尼、色瑞替尼、恩曲替尼和劳拉替尼。
ROS1 重排对 TKI 的耐药性在克唑替尼时代已被广泛研究,但对最近批准的 I 型 ROS1 TKI(即恩曲替尼和劳拉替尼)的耐药模式又开始出现。临床上已在克唑替尼耐药的肿瘤中发现了 ROS1 激酶结构域外显子的二次突变。最常见的是,它不仅会导致对克唑替尼的耐药性,还会导致对其他抑制剂的耐药性,包括恩曲替尼和劳拉替尼。已经观察到下一代 ROS1/TRK/ALK 抑制剂有希望的临床前活性(和)和早期临床疗效。劳拉替尼可以靶向与克唑替尼耐药相关的其他激酶突变,包括 和 ,后者也对恩曲替尼敏感。 II 型 ROS1 抑制剂,如卡博替尼,可能对接受 I 型 ROS1 抑制剂治疗的肿瘤患者有效,但这种“药物类别转换”策略需要进一步研究。
对 ROS1-TKI 脱靶耐药的罕见机制包括上皮间质转化; SCLC过渡; KRAS、NRAS、BRAF 或 KIT 突变; MET扩增;发信号。
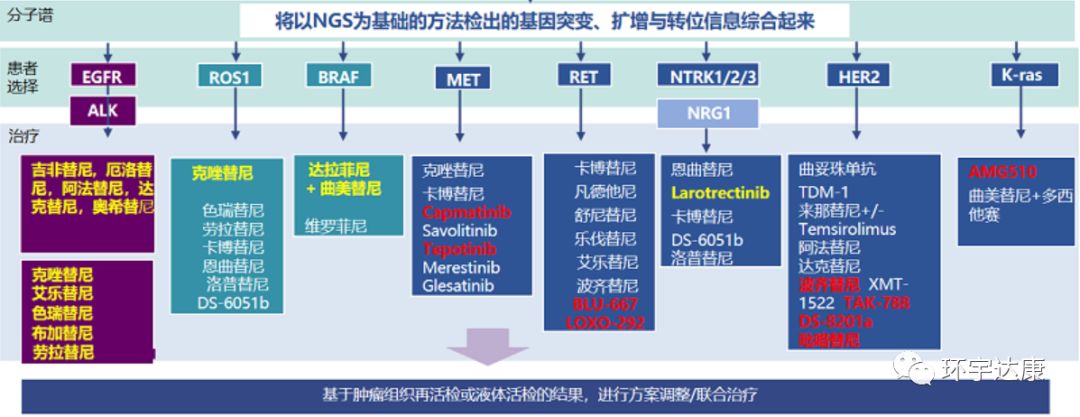
如果患者在治疗过程中出现耐药症状,需要通过基于NGS的检测方法,尽快从上述耐药机制中找出原因,选择相应的靶向药物和药物后续治疗的治疗方案。
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
