

欢迎光临吉康旅!
罕见病又被称为"孤儿病",目前,国际确认的罕见病种类共计5000~6000种,约占人类疾病的10%。各国对罕见病认定的标准存在一定差异,与其罕见病药物研发的激励政策及罕见病诊疗费用的覆盖范围有关。

由于我国人口基数大,罕见病患者在我国约有2000万人,根据世界卫生组织报道,约有80%的罕见病是由于遗传引起的,约有50%的罕见病在出生时或者儿童期即可发病。罕见病进展迅速,死亡率高,但却仅有约1%的罕见病有有效治疗药物。
这是因为罕见病发病率极低,一方面生物制药企业可以得到的研究资源极其有限,研究起来非常不容易;另一方面研究罕见病药物的投入多,经济利益小,所以制药企业对于罕见病药物的研发少之又少。
2018年5月22日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病》目录,涉及白化病、血友病、特发性肺动脉高压、肝豆病等在内的121种疾病。

据媒体报道,作为国家儿童医学中心,复旦大学附属儿科医院2017年成立全国首个儿童疑难罕见病门诊,根据统计,从2018年6月至2019年2月,罕见病门诊量达3016人次。2018年就诊人数前五名的罕见病门诊为血友病、先天性脊柱侧弯、结节性硬化、进行性肌营养不良和肝豆状核变性(肝豆病)。
所以,其实罕见病并没有我们想的那么"罕见",甚至像血友病、肝豆病等一些疾病还是我们经常听到的。
由于多数罕见病都是由于遗传引起的,所以想要防治或者减少罕见病,需要从根本上出发,专家提醒,必须构建新生儿疾病筛查网络,健全孕前产前检查和疾病筛查制度,努力降低包括罕见病在内的新生儿出生缺陷的发生率。另外,要做到早发现、早诊断、早治疗。
目前,国内罕见病用药较为缺乏,只有个别疾病有药物可用。如特发性肺动脉高压、特发性肺纤维化、肝豆病等。
特发性肺动脉高压
肺动脉高压被称为"心肺血管系统的癌症",患者由于缺氧导致手指和嘴唇呈蓝紫色,因此患者也被称为"蓝嘴唇",很多患者甚至不能大声唱歌、不能奔跑、不能情绪激动。
其实,肺动脉高压并不罕见,先天性心脏病、左心衰竭、瓣膜病、慢阻肺、风湿免疫系统疾病以及家族遗传等疾病都会引发肺动脉高压。只有特发性肺动脉高压(发病原因不明)才是罕见病。治疗肺动脉高压的药物相对较多,其中波生坦片等亦可用于特发性肺动脉高压。
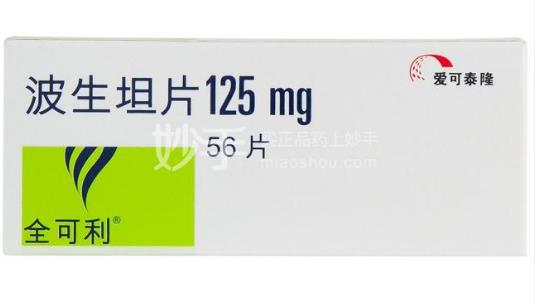
商品名:全可利
通用名:波生坦片
价格:4032元/盒(125mg*56片)
适应症:用于治疗WHO功能分级II级-IV级的肺动脉高压(PAH)(WHO第1组)的患者,以改善患者的运动能力和减少临床恶化。支持本品有效性的研究主要包括WHO功能分级II级-IV级的特发性或遗传性PAH(60%)、与结缔组织病相关的PAH(21%)及与左向右分流先天性心脏病相关的PAH(18%)患者。
特发性肺纤维化
特发性肺纤维化患者的肺被称为"丝瓜瓤",由于其肺组织上形成疤痕,失去弹性,变得像坚韧的丝瓜瓤一般。有些人一旦患病,病情会迅速恶化。有研究显示,超过50%的特发性肺纤维化患者往往看过3位医生后才能被确诊病情,诊断延误长达5年。有一半患者确诊后在2~3年内死亡,5年生存率低于30%,这比很多癌症的生存率都低。
临床上常用语治疗特发性肺纤维化的药物有乙磺酸尼达尼布软胶囊、吡非尼酮胶囊等。

商品名:艾思瑞
通用名:吡非尼酮胶囊
价格:819元/盒(100mg*54粒)
适应症:用于轻、中度特发性肺间质纤维化。
肝豆病
肝豆病全名为肝豆状核变性,又称威尔逊病,是少数可治疗的神经遗传病之一,不具有传染性,因该病铜代谢失常,患者又被称为"铜娃娃"。根据流行病学调查,肝豆病患病率为三万分之一。中国、日本、印度、美国发病率较高。
部分肝豆病患者会表现出肝功能异常,容易被当成肝炎来治疗。为此,不少患者被耽误了最佳治疗时机。但实际上,相较其他罕见病,肝豆病如果得到及时治疗,生存率是相对较高的,据了解,经过及时、正规的治疗,肝豆病患者的生存期远超10年,大多数患者的寿命甚至可近似正常人。

商品名:信谊
通用名:青霉胺片
价格:184元/盒(0.125g*100片)
适应症:1、用于治疗重金属中毒、肝豆状核变性(Wilson病)。 2、也用于其他药物治疗无效的严重活动性类风湿关节炎。

目前,中国罕见病领域的快速发展,已得到了国际社会的认可。但是,一个不争的事实是,当前在全国范围,甚至世界范围内,不仅治疗罕见病的药物稀缺,能够有罕见病诊疗能力的医生同样极为稀缺,所以全国甚至全球范围内对于罕见病的防治,依然面临严峻的考验,要战胜罕见病,仍需要企业、医生、患者共同努力。
想了解更多药品相关信息可查阅妙手医生商城或关注@妙手医生新特药 向专业药师进行咨询。
也许你还想看

免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
