

欢迎光临吉康旅!
/,一种突变特异性的第三代EGFR TKI,正在成为EGFR突变肺癌的一线治疗药物,但患者体内不可避免地会产生耐药性。我们在诱导肺腺癌的转基因小鼠模型中模拟了对奥希替尼的获得性耐药,发现它主要通过 EGFR 或 /Q 的继发性突变介导。对来自患者的循环游离 DNA 数据的分析表明,/V 突变几乎总是发生在驱动突变的背景下。在小鼠中进行的治疗测试表明,厄洛替尼和阿法替尼均能导致对奥希替尼耐药的肿瘤消退,而只有阿法替尼对突变肿瘤有效。一线奥希替尼联合厄洛替尼治疗可防止继发性 EGFR 突变的出现。
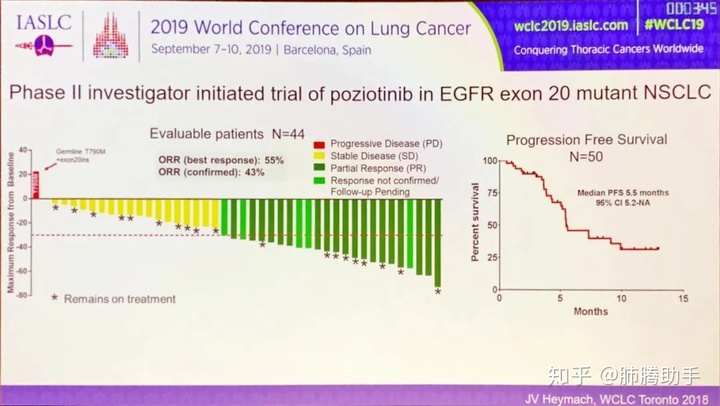
一线特瑞莎/奥希替尼的中位无进展生存期为 19 个月,总生存期为 39 个月,优于其他 TKI 观察到的结果。然而,患者并未治愈,需要更好地了解对奥希替尼的获得性耐药性。在这里,使用 EGFR 突变体 LUAD 的 GEMM,我们表明对一线奥希替尼的耐药性可能是由于 EGFR 中获得性和突变的 EGFR。这是在体内一线奥希替尼耐药模型中首次报道的获得 EGFR 突变的突变。来自一线奥希替尼试验的数据显示,在获得性耐药的情况下,分别有 7% 和 2% 的病例检测到 α 和 β 突变。在我们的鼠标模型中,与更均匀的突变分布相比,在这些患者样本中观察到的突变频率更高(具有多个驱动突变)和突变可能反映了耐药突变等位基因的特异性。事实上,我们对 EGFR 突变肺癌患者的分析清楚地表明,该突变在肿瘤亚群中占主导地位,并且 β-α 突变通常仅存在于 β-α 的情况下,这与少数已发表的文献形成鲜明对比。 /V的突变。为什么 /V 突变更有可能在患者的突变背景中发现仍有待确定。这可能是由于不同突变组合的稳定性不同或其他生化特性(例如它们的激酶活性或二聚化特性)的差异。有趣的是,我们研究和其他研究的体外数据表明,携带/V突变的细胞可以存在并对奥希替尼产生耐药性,这表明体内自发产生的耐药机制可能与观察到的机制有些不同。体外。
取代导致对特瑞莎/奥希替尼耐药的机制很简单,因为它去除了与奥希替尼共价反应的侧链硫醇。使用基于分子动力学的能量最小化获得的模型提供了一些关于 /Q 突变如何影响药物结合的见解。L718 侧链与奥希替尼结合的野生型 EGFR TKD (PDB ID 4ZAU) 晶体结构中的药物苯环接触。因此,正如其他人所指出的,用 V 或 Q 替换 L718 可能会改变奥希替尼结合的模式或方向。我们通过模拟突变 EGFR TKD 与奥希替尼、厄洛替尼和阿法替尼的非共价复合物证实了这一点。虽然奥希替尼在能量最小化的 TKD 复合物中(718 位亮氨酸)的位姿保持不变,突变的范德华相互作用消失,并与谷氨酰胺侧链的空间位阻发生冲突。突变。这些效应在模型中通过将奥希替尼重新定位在药物结合位点进行补偿,这可能会将其与 C797 的共价反应定向在次优方向 - 正如之前关于 /EGFR 的辩论中一样。奥希替尼的结合能也降低了,这表明在共价反应之前“遇到复合物”的亲和力可能会降低——这也会降低疗效。预计相同的突变对厄洛替尼结合能和方向具有相似的影响。在这种情况下,缺乏任何共价方面表明观察到的抗性一定是由于结合减弱所致。事实上,L718在EGFR/厄洛替尼晶体结构中直接接触厄洛替尼。最后,/Q 突变对阿法替尼结合能的影响仅为奥希替尼计算结果的 80% 左右,表明它们对阿法替尼共价反应的停留时间影响较小。此外,阿法替尼的方向似乎受突变的影响较小,虽然与 L718 的相互作用在变体中丢失,但其结合方向与 Q718 侧链之间没有冲突。
通过在编辑过的人类 EGFR 突变 LUAD 细胞系中进行的实验——模拟在存在低频亚克隆突变的情况下获得耐药性——我们表明,亚克隆/Q 突变赋予了对特瑞莎/奥希替尼的耐药性。然而,该突变对较高剂量的奥希替尼仍保持一定的敏感性,并且其在体内的频率随着奥希替尼的较高剂量而降低。在患者肿瘤活检报告中显示突变的仅有的三份报告也与 , 在我们的一只小鼠和获得 /Q 突变的编辑 PC9-VanR 细胞中复制。由 L718 突变引起的 EGFR 不稳定可能会阻止它们在存在的情况下发生。
/ 现在是晚期 EGFR 突变肿瘤的一线疗法,因此制定克服耐药性的策略至关重要。我们的研究结果证实了体内突变肿瘤对厄洛替尼的敏感性,正如几项体外研究和病例报告所表明的那样,但 /Q 突变肿瘤对厄洛替尼不敏感。如先前体外研究中所见,阿法替尼在所有轴承或突变肿瘤中引起肿瘤消退。这些数据与我们的报告一致,即患者通过在肿瘤中获得和突变而对一线奥希替尼产生耐药性,并且其疾病经阿法替尼稳定,类似于最近的两例二线奥希替尼阿法替尼病例报告+(15)和+突变肿瘤。基于其临床疗效,阿法替尼目前已获得FDA批准用于治疗具有突变的肿瘤,
获得性 EGFR 和 Kras 突变解释了此处描述的几乎所有特蕾莎/奥希替尼耐药 GEMM 肿瘤病例。然而,已在患者中观察到其他耐药机制,总体而言,我们发现 EGFR 的继发突变在我们的小鼠模型中比在奥希替尼耐药的人类肿瘤中更常见。这可能反映了 GEMM 肿瘤的基因组复杂性降低以及突变 EGFR 转基因的表达由多西环素持续驱动的事实,这可能导致更大的靶向抗性倾向。因此,应该使用其他模型来进一步发现不依赖 EGFR 的耐药机制并评估克服它们的治疗方法。尽管如此,我们的研究为 EGFR 依赖(和近端)对奥希替尼耐药的机制提供了重要见解,并强调了奥希替尼耐药的复杂性和异质性。我们的研究结果建议的临床研究的两个关键途径是:评估序贯 TKI 治疗,包括奥希替尼,其次是 2 个第二代 TKI,如阿法替尼或达克替尼,以及一线 TKI 组合评估。
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
