

欢迎光临吉康旅!
人体内糖原的贮存或消耗是一个受激素及底物控制的过程。通过调节参加合成及降解过程的酶的活性,机体的糖原代谢和血糖水准得到恰当的控制。糖原的不正常代谢,表现为糖原蓄积症,其原因常是由于缺乏有关的酶。例如,葡萄糖-6-磷酸酶缺乏的患者,肝及肾含有较多量结构正常的糖原,临床症状为肝肿大、极度低血糖、高脂血、高尿酸血、酮中毒以及生长停滞等。你好!
人体内糖原的贮存或消耗是一个受激素及底物控制的过程。通过调节参加合成及降解过程的酶的活性,机体的糖原代谢和血糖水准得到恰当的控制。糖原的不正常代谢,表现为糖原蓄积症,其原因常是由于缺乏有关的酶。例如,葡萄糖-6-磷酸酶缺乏的患者,肝及肾含有较多量结构正常的糖原,临床症状为肝肿大、极度低血糖、高脂血、高尿酸血、酮中毒以及生长停滞等。 采纳我 我绝对没参考
记得给问豆啊!
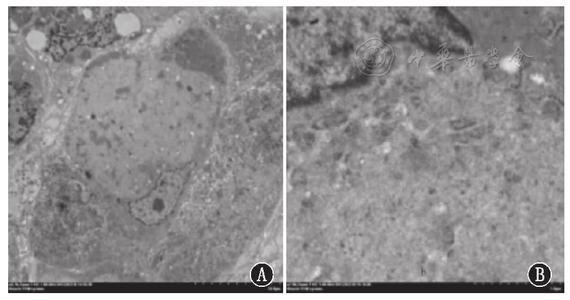
≒ompe’s disease)也称为糖原累积病II型(glycogenosis type II)或酸性麦芽糖酶缺乏症(acid maltase deficiency disease),是一种常染色体隐性遗传疾病,属于溶酶体贮积症(lysosomal storage disease)范畴。本病于1932年由Pompe首次报道,是糖原累积病中最常见的类型之一,有文献报道估计在中国人群中的发病率为1/50000,荷兰为1/40000,澳大利亚为1/146000。Pompe病由酸性a糖苷酶(GAA,也称为酸性麦芽糖酶)缺乏所致。其编码基因定位于17q25.2-q25.3,基因缺陷有点突变、剪切突变以及片段缺失等,目前已发现的突变超过200个,部分突变与特殊的临床表型相关联,比如c.-32-13T-G突变与晚发型Pompe病相关。GAA存在于人体内所有组织,在细胞内位于溶酶体,能将α-1 ,4糖苷键和α-1 ,6 糖苷键分解为葡萄糖分子。当GAA活性降低到一定水平时,溶酶体糖原开始出现堆积,此阈值水平可因器官不同而异。
本病男女均可罹患,部分学者推荐根据起病年龄和临床特点分为三型,即经典婴儿型、非经典婴儿型和晚发型(包括儿童型和成人型)。经典婴儿型在出生后数周或数月出现全身肌张力低下、无力,呈“软瘫婴儿”样表现,多见巨舌、心脏扩大和肝脏肿大,常于1岁之内死于心功能和肺功能衰竭。非经典婴儿型在2岁内起病,骨骼肌、心肌和肝脏受累程度较经典型轻。儿童型以骨骼肌受累多见,肝脏肿大少见,心肌通常不受累。患儿运动发育迟缓,易摔倒,部分有腓肠肌肥大。呼吸肌早期即受累,
呼吸衰竭是成年后常见的致死原因。成人型起病于18岁之后,也有更晚发病者,以慢性进展性肌病为主要表现,不伴脏器肿大,主要累积躯干和近端肌,呼吸肌也可选择性受累,容易被误诊为肢带型肌营养不良症或多发性肌炎。所有类型Pompe病的血清CK均增高,电生理均呈现肌源性损害。空泡性肌病为Pompe病的重要病理特点,组织生化提示细胞中的溶酶体内缺乏酸性麦芽糖酶。
酶替代治疗(ERT)是本病的重要治疗前景,目前美国FDA已批准MYOZYME?治疗本病你说呢...
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
