

欢迎光临吉康旅!
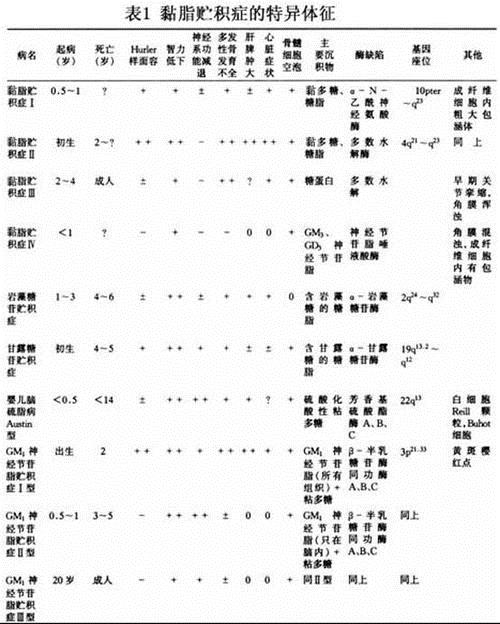
症状,1、粘多糖贮积症I型(mucopolysaccharidosis),又称Hurler综合征、承雷病。AR。为溶酶体a-L-艾杜糖醛酸酶缺乏,发病率1/10万新生儿。主征;进行性发育迟缓,体矮,智力低下,舟状头,颈短,面容粗陋,有角膜混浊,关节僵硬,活动受限,驼背。其他:视网膜色素沉着,耳聋,胸廊畸形,肝脾肿大,腹大,心脏瓣膜缺损和动脉硬化等。粘多糖筛查阳性,根据酶活性测定可检出携带者。预后差,多于儿童期死亡。,2、粘多糖贮积症II型(mucopolysaccharidosis),又称Hunter综合征。XR,无女性患者,发病率为6/10万。为艾杜糖醛酸硫酸酯酶缺乏。主征:与I型相似,但无角膜混浊,背不驼。其他:多毛,进行性耳聋;肌肉痉挛,行为莽撞。10岁后2/3患者有惊厥发作。生化诊断如I型,主要根据临床表现相鉴别。重型者多在青春期前死亡。,诊断,,,名称,代号,酶缺陷,酶学测定样品,生化改变,遗传特性,,,Hurler,Scheie综合征,MPSⅠ,α-L-艾杜糖酸苷酶,成纤维细胞、白细胞、组织、羊水细胞,尿和组织中DS、HS增多,成纤维细胞中DS增加,常染色体隐 性,,,Hunter综合征,MPSⅡ,艾杜糖醛酸硫酯酶,血清、成纤维细胞、白细胞、组织、羊水、羊水细胞,同上,X连隐性,,,Sanfilippo综合征A,MPSⅢA,HS-N-硫酸酯酶(硫酰胺酶),成纤维细胞、白细胞、组织、羊水细胞,HS在尿中和组织中增多、DS在成纤维细胞中增多,常染色体隐性,,,Sanfilippo综合征B,MPSⅢB,α-N乙酰葡萄胺苷,血清、成纤维细胞、白细胞、组织、羊水细胞,HS出现于尿中,同上,,,Sanfilippo综合征C,MPSⅢC,乙酰基转移酶,成纤维细胞,HS出现于尿中,同上,,,Morquio综合征,MPSⅣ,N-乙酰半乳糖胺-6-硫酸酯酶,成纤维细胞,KS和CS出现于尿中,同上,,,Morquio综合征,,β-半乳糖甘酶,成纤维细胞,KS出现于尿中,,,,Maroteaux-Lamy综合征,MPSⅥ,N-乙酰半乳糖胺4-硫酸酯酶(芳香硫酸酯酶B),成纤维细胞、白细胞、组织、羊水细胞,DS出现于尿中,同上,,,β-葡糖醛酸苷酶缺乏症无名疾病,MPSⅦ,β-葡糖醛酸苷酶,血清、成纤维细胞、白细胞、羊水细胞,DS、HS(±)出现于尿中,同上,,,无名疾病,MPSⅧ,N-乙酰葡糖胺6-硫酸酯酶,成纤维细胞,KS和HS(±)出现于尿中,同上,,,注:MPS-粘多糖沉积症DS-磷酸皮肤素,HS-硫酸乙酰肝素CD-硫酸软骨素,KS-硫酸角质素,遗传性粘多糖沉积症约占出生婴儿的1/30000。
粘多糖病是一组少见的先天性遗传疾病主要因降解粘多糖(现称糖氨聚糖)所需的溶酶体水解酶的缺陷,致使组织内有大量粘多糖蓄积,造成骨骼发育障碍、肝脾肿大、智力迟钝和尿中粘多糖类排出增多。粘多糖病I(H)型患者面容丑陋,形似中国古建筑屋檐下天沟(承)上的怪物,故也有承病之称。患者中男性多于女性,多见于近亲结婚者的后代,多有家族史。无特效治疗,只有对症和支持疗法。因酶缺陷的类型不同,预后不一。一般情况下,患儿多于出生1年后发病,10岁左右死亡,但有的病人可存活到50多岁。
粘多糖病是一组少见的先天性遗传疾病主要因降解粘多糖(现称糖氨聚糖)所需的溶酶体水解酶的缺陷,致使组织内有大量粘多糖蓄积,造成骨骼发育障碍、肝脾肿大、智力迟钝和尿中粘多糖类排出增多。粘多糖病I(H)型患者面容丑陋,形似中国古建筑屋檐下天沟(承)上的怪物,故也有承病之称。患者中男性多于女性,多见于近亲结婚者的后代,多有家族史。无特效治疗,只有对症和支持疗法。因酶缺陷的类型不同,预后不一。一般情况下,患儿多于出生1年后发病,10岁左右死亡,但有的病人可存活到50多岁。
一组少见的先天性遗传疾病主要因降解粘多糖(现称糖氨聚糖)所需的溶酶体水解酶的缺陷,致使组织内有大量粘多糖蓄积,造成骨骼发育障碍、肝脾肿大、智力迟钝和尿中粘多糖类排出增多。粘多糖病I(H)型患者面容丑陋,形似中国古建筑屋檐下天沟(承)上的怪物,故也有承病之称。
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。










微信扫码◀
免费咨询电话
